Research in our lab focuses on genetic variation and plasticity. We try to identify molecular pathways underlying phenotypic differences among genotypes, particularly differences in how each genotype reacts to dynamic environments. Our goals are to improve crop performance predictions, learn about forces that shape the evolutionary histories of natural populations, and identify critical systems that limit plant responses to climate change.
We approach these goals with various tools, including statistics, quantitative genetics, gene network and ecophysiological models, bioinformatics, and lab experiments. Most of our projects use gene expression analysis to probe molecular variation involved in particular gene networks and try to link gene expression traits to variation in development or physiology.
Here is an overview of our current work:
Interaction of gene signaling networks and physiology in flowering time control
Plants in the field face multiple simultaneous environmental signals that can occur at unpredictable times during development. Two complementary approaches are used to predict developmental responses such as flowering to such environmental signals: physiology-based “crop models”, and gene network-based models of gene interactions. Crop models are based on simple developmental rules and excel at predicting responses to continuously variable environments (Donohue et al 2014). But these developmental rules are not closely tied to molecular mechanisms, and so have little power to extrapolate to new genotypes. Gene network models are useful for identifying the set of gene interactions necessary to explain variation among genotypes, but few are incorporate dynamic environmental inputs.
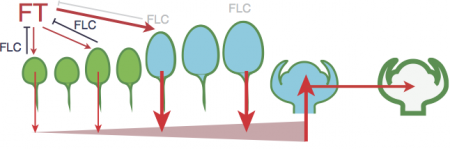
We aim to bridge these two approaches by identifying correlations between the expression dynamics of key integrator genes in flowering time pathways and the dynamics of developmental variables in well-parameterized crop models. Our hypothesis is that the quantitative behavior of certain signaling pathways is dependent on the physiological history and morphology of the plant. Gene networks that integrate signals from multiple tissues (such as daylength signals from multiple leaves) provide a mechanism for temporal averaging of environmental signals if those tissues were produced under different environments and maintain different autonomous gene network states. As a first step, we are testing if this hypothesis can reconcile the quantitative dynamics of responses to winter chilling, or vernalization, described by a successful crop model for the model plant Arabidopsis thaliana (Wilczek et al 2009), with the molecular mechanisms known in great detail in this species. To do so, we have measuring the expression of key genes in multiple tissues exposed to different environmental histories. We aim to further explore these ideas with respect to other environmental signals in other species such as wheat.
Bayesian statistical methods for quantitative genetics of gene expression and other high-dimensional traits
Genomic assays of gene expression offer the possibility of studying evolution in molecular traits such as gene networks or development. However, the immense number of gene expression traits is too large for traditional analytical approaches used in quantitative genetics to be useful. In collaboration with Sayan Mukherjee in the Statistics Department at Duke, I developed a statistical model called Bayesian Sparse Factor Analysis of Genetic Covariance Matrices, or BSFG, suitable for estimating genetic correlations at the transcriptome-scale (Runcie and Mukherjee, Genetics, 2013). This model is motivated by the idea that gene expression traits are modular, and therefore we can increase our power by looking explicitly for highly correlated groups of genes. Our model produces robust estimates of genetic covariance matrices (G-matrix) for thousands of traits at once. At the same time, our model identifies highly correlated gene sets that may be signatures of variation in important molecular and developmental traits and quantifies their heritability. This model is implemented in freely available software.
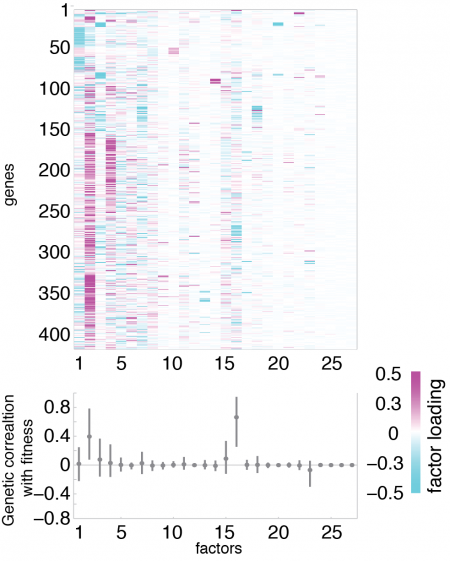
Current projects involve extensions of this modeling framework to high-dimensional QTL and GWAS applications, to robust analysis of gene expression responses in complex designed experiments, and to incorporating prior information on gene sets such as gene function, co-expression, or tissue specificity.
A talk describing the method is published here: https://doi.org/10.6084/m9.figshare.4906322.v1
Software is available here: https://github.com/deruncie/SparseFactorMixedModel
Gene expression traits underlying adaptation to high elevation in maize landraces
After domestication from the wild progenitor teosinte, maize populations adapted to high elevations multiple times across the world. C4 plants are uncommon in environmental conditions typical of high elevation environments, and so this adaptation likely required multiple changes in physiology, phenology and morphology particularly for the tolerance of cold, water stress and UV. In collaboration with Jeffrey Ross-Ibarra, Matthew Hufford, Ruairidh Sawyers, and Sherry Flint-Garcia, we are studying the population and functional genomics of this adaptation. Our lab’s contribution to this project is to use gene expression assays to identify candidate genes and pathways relevant to local adaptation. Our approach is two-fold: First, we are studying the gene expression response to cold temperatures across multiple tissues in high and low elevation genotypes. Second, we are looking for population genetic signatures of selection on gene expression profiles between low and high elevation populations of maize using genome-wide RNAseq-based allele-specific expression assays.
See news report here.
Convergent evolution of critical photoperiod shifts in a flowering time gene network
The timing of flowering in response to photoperiod cues frequently shows strong evidence for local adaptation across wild populations inhabiting different environments such as clines in latitude or elevation. Studies in Arabidopsis thaliana have shown that several genetic pathways can control the response of flowering to photoperiod. The overall topology of flowering time pathways are largely conserved across diverse species. In collaboration with Benjamin Blackman, Jason Sexton and Akiko Satake, we are studying whether parallel shifts in the critical photoperiod that can trigger flowering along multiple altitudinal clines are the result of perturbations to the same genetic pathways.
To test this hypothesis, we are collecting fine-scaled data on the expression of key nodes in flowering time and circadian pathways for contrasting high and low elevation genotypes across several parallel clines in multiple temperature and photoperiod regimes. We will use these data to parameterize computational models of the genetic control of flowering time, and then test model predictions by growing specific genotypes in the field.
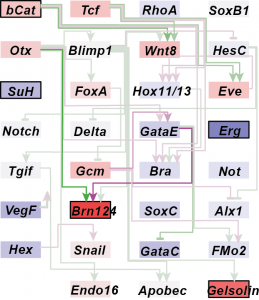
Genetic variation and plasticity in a developmental gene regulatory network
Regulatory genes and signaling molecules operate in gene networks to control development and responses to the environment. However, we know little about how genetic variation operates through gene networks to produce phenotypic variation in natural populations. A major focus of my dissertation research with Greg Wray at Duke University was to test how a developmental gene network that drives early morphogenesis in sea urchins shapes phenotypic variation in different environments. Using a quantitative genetics approach, I measured gene expression variation in this network to address the question: Do gene networks influence the expression of genetic variation under realistic environmental stresses? I documented substantial genetic variation and plasticity in the expression of critically important developmental genes in the network, and significant genetic correlations between the expression profiles of directly interacting genes (Runcie et al Mol Ecol. 2012, Garfield, Runcie et al PloS Biology 2013). Surprisingly, gene expression responses to high temperatures did not follow the network topology. This demonstrates that the gene network function was effectively buffered across a moderate temperature range. Mechanisms that buffer gene network outputs are, therefore, critical for understanding gene-trait relationships in different environments.